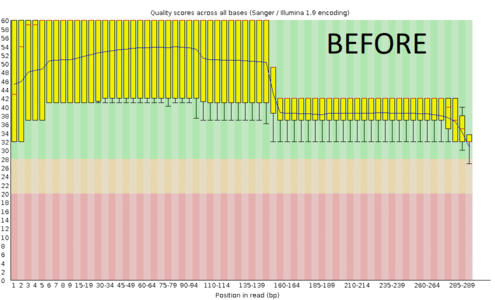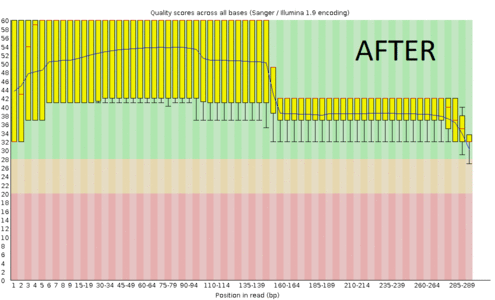
Creating consensus fasta using iupac codes
Published:
Fasta files can be generated from vcf calls. There are two ways of doing that: (1) concatenate snips together (this can be done using either variants only or calling monomarphic (hom ref) variants as well and concatenating them too); (2) use reference genome as a backbone and incorporate variants into the reference. To incorporate information about heterozygotes, IUPAC substitution codes can be used. Here is a collection of scripts available:
Using fasta reference
- the best and most convenient way: (allows to choose a sample from multiVCF and BED mask)
bcftools consensus --iupac-codes --sample --mark - vcf2fasta.py - phased only
- For GATK fans: FastaAlternateReferenceMaker.
No reference
- seqTools vcf2fasta
- VCF2FASTA_IUPACcoding.r - something complicated that can be adapted to your needs.
- VCF-to-Tab_to_Fasta_IUPAC_Converter.py - this code can be used on BSNP file. Keep in mind, that if you need to filter out transitions (in case of aDNA) and you use BSNP output, you need a reference to see wich substitutions are transitions. Thus, additional masking is needed.
- freebayes_vcf2fa.py - freebayes only