
You May Also Enjoy
Dear Aspiring Artist
3 minute read
Published:
Copypaste from http://www.hrgiger.com/
Creating consensus fasta using iupac codes
less than 1 minute read
Published:
Fasta files can be generated from vcf calls. There are two ways of doing that: (1) concatenate snips together (this can be done using either variants only or calling monomarphic (hom ref) variants as well and concatenating them too); (2) use reference genome as a backbone and incorporate variants into the reference. To incorporate information about heterozygotes, IUPAC substitution codes can be used. Here is a collection of scripts available:
Tools to deal with aDNA damage
less than 1 minute read
Published:
- mapDamage –rescale option changes quality scores on bam files
- PMDtools - filters out contaminant reads based on deamination profiles AND adjust scores too. Works on SAM files.
- ATLAS toolset can do BQSR
- AntCaller - genotype caller for ancient genomes. Works better than GATK according to the paper.
- SpAl - estimate the proportion of spurious alignments in ancient DNA. Works with any species, as long as there is data on variation in the genome.
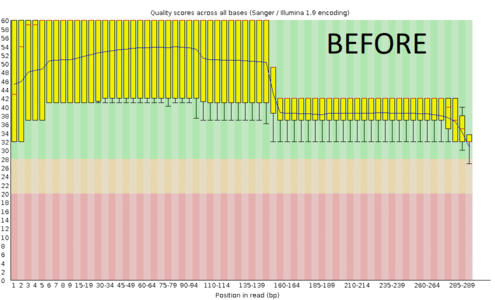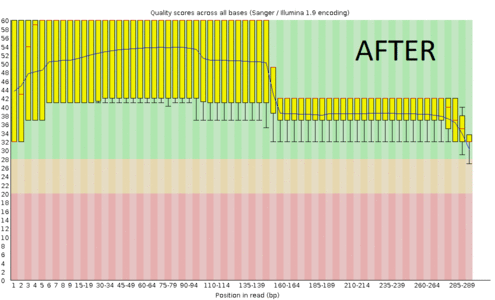
Should we BQSR ancient genomes?
1 minute read
Published:
A regular protocol for variant discovery pipeline includes base call recalibration after an alignment to a reference genome. This is usually done by GATK. Recalibration accounts for the fact that quality scores sometimes can depend on a cluster density of the sequencing machine, length of the read, position of a bp in the read and other factors. When sequencing ancient DNA, all these parameters go out the window because of DNA post mortem damage (PMD). So should we recalibrate anyway?