Dear Aspiring Artist
Published:
Copypaste from http://www.hrgiger.com/

Published:
Copypaste from http://www.hrgiger.com/
Published:
Fasta files can be generated from vcf calls. There are two ways of doing that: (1) concatenate snips together (this can be done using either variants only or calling monomarphic (hom ref) variants as well and concatenating them too); (2) use reference genome as a backbone and incorporate variants into the reference. To incorporate information about heterozygotes, IUPAC substitution codes can be used. Here is a collection of scripts available:
Published:
Published:
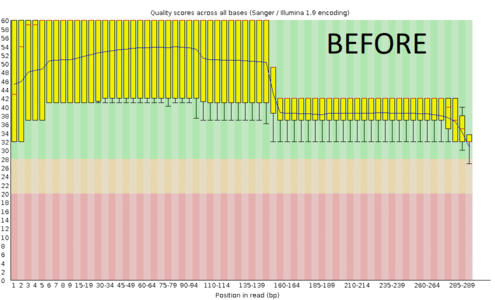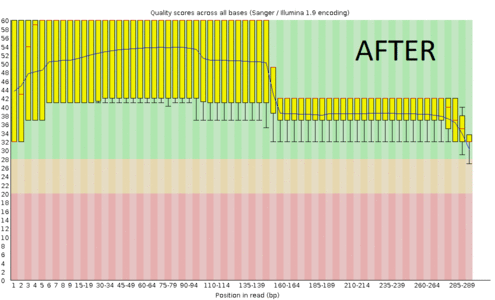
A regular protocol for variant discovery pipeline includes base call recalibration after an alignment to a reference genome. This is usually done by GATK. Recalibration accounts for the fact that quality scores sometimes can depend on a cluster density of the sequencing machine, length of the read, position of a bp in the read and other factors. When sequencing ancient DNA, all these parameters go out the window because of DNA post mortem damage (PMD). So should we recalibrate anyway?
Published:
f3 statistic was developed by Patterson and Reich to measure admixture between populations. It is widely used in ancient DNA studies and there are many packages out there which are able to estimate it for you.
Published:
There are several potential problems if you call SNPs from low coverage ancient samples: